Krankheitsmechanismen der Mastzellaktivierungserkrankungen (MCAD)

Seitenübersicht:
Für das bessere Verständnis sollte zuvor die Seite Mastzellerkrankungen > Normale Mastzellfunktion gelesen werden, auf der diese Seite aufbaut.
Zusammenfassung:
Mastzellen werden im Knochenmark aus Blutzellen bildenden Vorläuferzellen gebildet. Von dort wandern sie durch das Gewebe und reifen zu Mastzellen heran, um schliesslich in irgend einem Organ ihre Funktion zu erfüllen. Diese Funktion besteht darin, auf bestimmte Reize zu reagieren und Mediatoren (Botenstoffe) auszuschütten.
Irgendwann im Laufe des Lebens kann eine einzelne Vorläufer- oder Mastzelle durch eine Genmutation eine krankhafte Veränderung erfahren, derart, dass sie entweder daueraktiviert ist, oder viel leichter aktivierbar (überempfindlich) wird. In solchen Fällen handelt es sich folglich um eine im Laufe des Lebens erworbene Erkrankung, die nicht direkt vererbt wird. Erbliche Varianten kommen jedoch ebenfalls vor.
Der Aktivitätszustand der mutierten Mastzellen ist dauerhaft erhöht und dadurch nicht mehr regulierbar. Die Überaktivierung löst in den aktivierten Zellen folgende Reaktionen aus:
- Erleichterte Freisetzung und vermehrte Neusynthese von Mastzellmediatoren
- Aktive Wanderung durch das Gewebe
- Zelldifferenzierung (d.h. Spezialisierung auf bestimmte Funktionen)
- Langlebigkeit durch gestörte Apoptose (=programmierter Zelltod). Dass sie nicht mehr zum geplanten Zeitpunkt eliminiert werden, erhöht deren Anzahl.
- Bei bestimmten Mutationen ist auch eine verstärkte Vermehrung (Proliferation, Zellteilung) der Mastzellen zu beobachten. Dies führt zur Anreicherung krankhaft veränderter Mastzellen in den betroffenen Organen oder Geweben, was zu einer sonographisch sichtbaren Vergrösserung befallener Organe führen kann.
Sinn und Zweck der Freisetzung hunderter verschiedener Mastzellmediatoren ist es, mit diesen Botenstoffen biochemische Signale an andere Zellen weiter zu geben. Die umliegenden Zellen sollen informiert werden, was in der aktuellen Situation zu tun ist. Die Überaktivierung kann auf diese Weise zunächst lokal zu Symptomen führen, indem die umliegenden Zielzellen (normale Körperzellen) durch die ausgeschütteten Mediatoren in einen Ausnahmezustand versetzt werden.
Vor allem werden aber durch die ausgeschütteten Mediatoren auch die noch gesunden Mastzellen in der Umgebung angelockt und ebenfalls aktiviert (sekundäre Aktivierung), wodurch auch diese beginnen, Mediatoren auszuschütten. Dies aktiviert wiederum noch mehr Mastzellen in noch grösserem Umkreis. Ab einer gewissen Signalstärke können die Mediatoren bis ins Lymphsystem und in die Blutbahn gelangen. Dies mobilisiert und aktiviert wiederum noch mehr Mastzellen in noch grösserer Entfernung. Auf diese Weise kann sich der Krankheitszustand mit der Zeit auf den gesamten Körper ausbreiten. So kann eine lokale Mutation zu einer systemischen (=den gesamten Körper betreffenden) Erkrankung führen.
Bei sehr ungünstigen Verlaufsformen werden auch Nicht-Mastzell-Immunzellen mit stimuliert. Bei einer solchen immunsystemübergreifenden Erkrankung zeigt eine mastzellspezifische Medikation oft keinen Erfolg mehr.
Auf der vorhergehenden Seite wurde die Beschaffenheit und Funktion gesunder Mastzellen erklärt. Nun gehen wir auf die verschiedenen krankhaften Vorgänge ein, an denen Mastzellen beteiligt sind und bei denen Histamin und andere Mediatoren freigesetzt werden.
Kurze Einführung in die Grundlagen der Genetik
(Wer bereits Grundkenntnisse in Genetik hat, oder es gar nicht bis ins Detail verstehen möchte, kann dieses Kapitel überspringen.)
Was ist ein Gen?
Jeder Mensch besteht aus mehreren Billionen kleiner Zellen und in jeder einzelnen Zelle steckt eine vollständige Kopie des gesamten Erbguts mit etwa 30'000 verschiedenen Genen. In diesen Genen steckt die Erbinformation, welche die verschiedensten Eigenschaften und Mechanismen unseres Körpers bestimmt und von Generation zu Generation weiter vererbt wird. Gene sind einzelne Informationsabschnitte im Erbgut, von denen vereinfacht gesagt jeder Abschnitt die Information enthält, wie ein ganz bestimmtes Protein (=Eiweiss) aufgebaut sein muss. Genauer: In welcher Reihenfolge die Aminosäuren, aus denen die Proteine bestehen, kettenförmig aneinander gereiht werden müssen. Die Aminosäuren tragen unterschiedliche Ladungen, die lokal zu anziehenden und abstossenden Kräften führen, so dass sich der Proteinstrang zu einem Knäuel mit einer ganz bestimmten Form zusammenfaltet. Durch diesen bestimmten, individuellen Aufbau und durch die Ladungen auf seiner Oberfläche erhält jedes Protein eine ganz bestimmte Funktion. Einige Proteine sind Baustoffe (z.B. Haut und Bindegewebe aus Kollagen, Haare und Fingernägel aus Keratin), andere sind Enzyme. Enzyme sind eine Art "Maschinen", die durch ihre blosse Anwesenheit und ihre individuelle Struktur bewirken, dass an ihrer Oberfläche ganz bestimmte chemische Reaktionen ablaufen können, die sonst nicht ablaufen könnten. Z.B. die Diaminoxidase (DAO) und die Histamin-N-Methyltransferase (HNMT), welche Histamin abbauen können.

Gendefekte, Genvarianten, Polymorphismen
Fehler sind als Motor der Evolution überlebensnotwendig für die Art
Als kleine Randbemerkung sei noch erwähnt: Diese zufällig eingebauten Fehler entstehen nicht, weil die Natur nicht perfekt wäre und es nicht besser könnte. Sie sind von der Natur so "gewollt", denn nur durch ständige Veränderung kann Neues ausprobiert werden. Jede Art hat eine für sie ideale, artspezifische Fehlerrate beim Kopieren der Gene. Viren müssen rasend schnell mutieren, um unser Immunsystem austricksen können. Bei einem hoch komplexen und hoch entwickelten Organismus wie dem Menschen ist hingegen nur eine deutlich kleinere Fehlerrate sinnvoll. Dieser planlose "Erfindergeist" der Evolution ermöglicht Anpassung und Weiterentwicklung, sofern die Mutationen eine Eizelle oder ein Spermium betreffen und dadurch an Nachkommen vererbt werden können. Für das betroffene Einzelindividuum kann das zwar fatal sein, weil die allermeisten Mutationen sich nachteilig auswirken, aber die Art bzw. Population als ganzes profitiert davon, weil sich ein paar wenige dieser abweichenden Varianten als vorteilhafte Neuentwicklungen durchsetzen. Womöglich wird eine dieser Varianten sogar irgendwann als einzige in der Lage sein, bestimmte extreme Umweltbedingungen überleben zu können. Genetische Vielfalt (d.h. von möglichst vielen Genen sollten in jeder Population möglichst viele abweichende Varianten vorkommen) kann aus diesem Grunde wichtig sein für das Überleben einer Art.
Überall passieren Fehler. Beim Abschreiben von Text genauso wie beim Kopieren des Erbguts. Die Gene in einem Lebewesen kann man sich vorstellen als "Wörter" in einem "Buch" (Genom, gesamtes Erbgut), die aus vielen aneinander gereihten "Buchstaben" (Nukleotiden) bestehen. Auch beim Kopieren der Erbsubstanz in den Zellen werden manchmal zufällig falsche "Buchstaben" eingebaut, die vom Original abweichen. Die meisten dieser zufälligen "Tippfehler" (Genvarianten) ergeben allerdings Unsinn. Ein Beispiel zur Veranschaulichung: Ein zufälliger Tippfehler im Wort "Besen" wird wohl meistens eine Buchstabenfolge ohne Bedeutung ergeben, z.B. "Bxsen", was beim Buchkritiker mindestens einen schlechten Eindruck hinterlässt oder schlimmstenfalls sogar dazu führen kann, dass der gesamte Text keinen Sinn mehr ergibt. Manchmal entstehen so aber zufällig Wörter, die dem Text einen ganz neuen Sinn geben, z.B. "Beben", "Basen", "Bösen" oder "Busen", was je nach Kontext und Zielpublikum den Leser entweder verwirren oder aber dem Buch vielleicht sogar zu mehr Erfolg verhelfen kann.
, Mutation. Quelle: Wikipedia. Urheber: David Hall")
Weil alle Gene schon seit hunderttausenden von Jahren über unzählige Generationen hinweg immer wieder kopiert wurden, gibt es heute von den meisten Genen mehrere verschiedene Varianten, was zur Folge hat, dass sich die einzelnen Individuen voneinander unterscheiden. Auch vom DAO- und vom HNMT-Gen hat man in Studien mehrere verschiedene Varianten gefunden, von denen nicht alle das Histamin gleich schnell abbauen können. Dass in der Bevölkerung von einem Gen mehrere Varianten existieren, nennt man in der Fachsprache Polymorphismus (von poly = viel und morph = förmig) oder auch "Single Nucleotid Polymorphism" (SNP) für eine Variante, die durch Austausch eines einzelnen Bausteins ("Buchstabens") entstanden ist (siehe Abbildung).
Der Begriff "Gendefekt" ist zwar umgangssprachlich geläufiger und leichter verständlich als der Fachbegriff Polymorphismus, jedoch auch ethisch problematisch und politisch wie auch sachlich nicht korrekt: Ob eine Genvariante sich vorteilhaft oder nachteilig auswirkt, hängt von der Umwelt ab, in der sich dieses Gen (bzw. der Organismus) behaupten muss. Eine Genvariante an sich kann deshalb nicht "gut" oder "schlecht", "gesund" oder "defekt" sein.
Ein eindrückliches Beispiel hierfür ist die Sichelzellenanämie. In den meisten Regionen stellt diese Erbkrankheit, die zu sichelförmig verkrüppelten roten Blutkörperchen führt, einen Überlebensnachteil dar, so dass die Krankheit entsprechend sehr selten vorkommt. In einigen Malariagebieten ist die Sichelzellenanämie aber weit verbreitet, weil die veränderten Blutkörperchen nicht vom Malariaerreger befallen werden können. Die Träger dieser Mutation sind vor Malaria geschützt und haben deshalb unter diesen Umweltbedingungen die besseren Überlebenschancen als die Träger der "normalen" Genvariante.
Die Laktose-Unverträglichkeit Erwachsener ist ebenfalls kein "Fehler", sondern hat einen biologischen Sinn: So soll verhindert werden, dass die älteren, selbständigeren Kinder zu Nahrungskonkurrenten der später geborenen und noch voll auf Muttermilch angewiesenen Kinder werden. Somit ist eigentlich das Vertragen von Milchzucker der "Fehler", der sich für einzelne Völkergruppen mit dieser Mutation aber irgendwann als vorteilhaft herausstellte, als diese lernten, wild lebende Säugetiere zu domestizieren und zu melken.
Der wertende Begriff "Gendefekt" ist deshalb eigentlich nicht korrekt, wird aber zwecks besserer Verständlichkeit dennoch oft verwendet.
"Entartete" Zellen nach Mutationen in Protoonkogenen
Der menschliche Körper besteht aus etwa 50 Billionen Zellen. In jeder einzelnen Zelle ist jeweils das gesamte Erbgut und damit der vollständige Bauplan des gesamten Körpers enthalten, der etwa 3.2 Milliarden einzelne "Buchstaben" (Nukleotide) umfasst. Darin enthalten sind etwa 25'000 Abschnitte, die eine bestimmte Bedeutung haben und die man Gene nennt. In diesem Erbgut entstehen, weil es so umfangreich ist, entsprechend viele Fehler, sei es spontan beim Kopieren, durch chemische Einflüsse (krebserregende oder erbgutverändernde Substanzen) oder durch energiereiche Strahlung (Röntgenstrahlen, Radioaktivität, UV-Licht der Sonne). Im Laufe unseres Lebens treten deshalb in unzähligen Zellen unseres Körpers solche Fehler auf. Ein Teil davon bleibt folgenlos, andere können aber dazu führen, dass das Gen, in dem die Mutation liegt, seine Funktion verliert oder verändert. Da nur eine einzelne von vielen Billionen Zellen betroffen ist, hat das zwar manchmal ungünstige bis fatale Folgen für die einzelne Zelle, aber nicht für den Körper als Ganzes. Für den Körper ist der Verlust oder die Fehlfunktion einer Einzelzelle vernachlässigbar. Es gibt aber ein paar verhängnisvolle Ausnahmen, wie wir gleich sehen werden:
Zu den Protoonkogenen (aus "proto" = vor, "onko" = Krebs" und "Gen" = ein Abschnitt im Erbgut) zählt man alle Gene, deren Ausfall zu einer Entartung der betroffenen Zelle in irgend einer Form führen würde, wenn eine funktionsverändernde Mutation in diesem Gen auftreten würde (meist eine ungehemmte Vermehrung). Dazu gehören insbesondere diejenigen Gene, die in irgendeiner Weise – direkt oder indirekt – am normalen Zellwachstum, an der Zellteilung oder an der Zelldifferenzierung beteiligt sind. Wenn ein solches Protoonkogen durch eine Mutation seine Funktion verloren hat, nennt man es Onkogen (Krebsgen). Die betroffene Zelle gerät ausser Kontrolle. Was dann passiert, hängt davon ab, was genau durch die Mutation kaputt gegangen ist. Die schlimmstmögliche Folge wäre, dass ein bösartiger Tumor oder Blutkrebs (Leukämie) entsteht und sich im Körper ausbreitet. Es gibt aber noch ganz andere, sehr spezielle Möglichkeiten, was dabei durcheinander geraten kann. Eine davon schauen wir uns gleich etwas genauer an:
Körperliche Ursachen von Mastzellaktivierungserkrankungen
Bei den körperlichen Ursachen auf molekularer Ebene handelt es sich um genetische Veränderungen, welche zu einer Aktivitätssteigerung der betroffenen Mastzellen führen.
Im Körper gibt es verschiedene Zelltypen, die auf unterschiedliche Aufgaben spezialisiert sind (so ähnlich wie Menschen mit unterschiedlichen Berufen, die zusammen eine Gemeinschaft bilden). Die Spezialisierung entsteht, indem nicht in allen Zellen alle Gene gleichermassen aktiv sind. Mastzellen haben vor allem die Aufgabe, Bedrohungen zu erkennen und darauf hin Botenstoffe auszuschütten. In den letzten Jahren hat man verschiedene Mutationen in verschiedenen Genen entdeckt, welche sich störend auf das Funktionieren der Mastzellen auswirken [Traina et al. 2012]. Wahrscheinlich muss sogar meistens eine Kombination mehrerer, erst unvollständig bekannter, Genmutationen vorliegen. In Frage kommen Veränderungen an Kinasen, Rezeptoren und anderen Proteinen der biochemischen Signalübertragungskette.
Somatische Mutationen, Klonalität
Mastzellmutationen sind fast immer somatisch. Somatisch bedeutet, dass die Mutation zufällig irgendwann im Laufe des Lebens in einer einzelnen Zelle auftritt und sich mit der Teilung dieser Zelle verbreitet, dass aber die Keimbahn (Spermien, Eizellen) nicht betroffen ist und dadurch die Mutation nicht vererbt wird. Meistens sind alle mutierten Zellen aus einer einzelnen mutierten Vorläuferzelle im Knochenmark hervorgegangen. Dies nennt man Klonalität oder (mono)klonale Zellen. Auch reifende oder ausgereifte Mastzellen irgendwo im Körper können mutieren.
Vererbbare Mutationen (Keimbahnmutationen)
Vererbbare Mutationen sind ebenfalls möglich. In diesem Fall tragen sämtliche Körperzellen die Mutation. Dadurch ist auch die Keimbahn (die der Fortpflanzung dienenden Geschlechtszellen: Spermien bzw. Eizellen) betroffen. Die Mutation wird an die Nachkommen weiter gegeben und die Krankheit tritt familiär gehäuft auf. Man weiss leider noch sehr wenig darüber, kann sie noch nicht mittels Routinediagnostik testen und entdeckt sie dem entsprechend sehr selten. [Pardanani 2012, S.403 und 405; Valent et al. 2012, S.215]
Verteilungsmuster mutierter Mastzellen
Typischerweise ist nur ein kleiner Teil der Mastzellen in einem Teil des Körpers betroffen. Sämtliche Organe und Gewebe können betroffen sein. Meistens sind mehrere Organsysteme betroffen.
Mastzellaktivierende Pathomechanismen
Es gibt wohl verschiedene molekulare Mechanismen, die eine Mastzellaktivierungserkrankung hervorrufen können. Hier stellen wir einen vor, der bereits erforscht ist:
Die Tyrosinkinase KIT, ein Stammzellfaktor-Rezeptor
![[Tyrosinkinase KIT]](https://de.wikipedia.org/w/index.php?title=Datei:Kit_ligand_2E9W.png&filetimestamp=20081008082335 "3D-Oberflächenmodell des Stammzellfaktors SCF (grün) mit seinem Rezeptor Tyrosinkinase KIT (blaugrau).")
Der wichtigste Wachstumsfaktor der Mastzellen ist ein Zytokin mit der Bezeichnung "Stammzellfaktor" ("stem cell factor", SCF). Der Stammzellfaktor steuert das Wachstum der Mastzelle, indem es an den Stammzellfaktor-Rezeptor bindet und diesen dadurch aktiviert. Dieser Rezeptor heisst Tyrosinkinase KIT (oder CD117, c-Kit) und steckt in der Zellmembran auf der Oberfläche der Zelle. [Yuzawa et al. 2007]
Wenn die Tyrosinkinase KIT durch den Stammzellfaktor aktiviert wird, schaltet dieses Signal mehrere Zellfunktionen ein: Die Zelle vermehrt sich, sie differenziert sich (d.h. spezialisiert sich auf bestimmte Funktionen), sie setzt Mediatoren frei (hauptsächlich Histamin), sie wandert durch das Gewebe und sie wird langlebiger.
Bestimmte Mutationen in einem bestimmten Bereich des KIT-Gens (in Exon 17) verändern jedoch den genetischen Bauplan so, dass eine verkrüppelte Tyrosinkinase KIT hergestellt wird, welche bereits die Form aufweist, die sie normalerweise erst dann einnehmen würde, wenn der Wachstumsfaktor angedockt hat. Diese Daueraktivierung hat zur Folge, dass der Zelle andauernd das Signal zum Wachstum gegeben wird, unabhängig davon ob der Wachstumsfaktor anwesend ist oder nicht. [Hartmann et al. 2009]
Bei Fällen mit Mastozytose findet man weitaus am häufigsten eine somatische D816V (Asp-816-Val) Punktmutation im Gen der Tyrosinkinase KIT. Laut Akin et al. tragen mehr als 90 % aller Erwachsenen mit systemischer Mastozytose diesen Gendefekt [Akin et al. 2010, S.1100]. In anderen Studien wird die Häufigkeit von D816V mit 29 bis 100 % der SM-Patienten angegeben [Traina et al. 2012].
Andere, noch weitgehend unerforschte Ursachen
Nebst dem Codon KIT D816V sind auch zahlreiche andere Mutationen in diesem Gen sowie in anderen Genen ("platelet derived growth factor alpha", Rezeptorproteine) identifiziert worden, die zu einer Daueraktivierung der betroffenen Mastzellen führen [Molderings et al. 2010, S.723; Pardanani 2012, S.403] oder die zumindest in Mastozytose-Patienten sehr häufig vorkommen, ansonsten aber noch nie gefunden wurden [Traina et al. 2012]. Offenbar hat die Mutation D816V alleine relativ milde Auswirkungen und führt erst in Kombination mit unbekannten weiteren Mutationen zu einer starken Aktivierung [Chaix et al. 2013]. Zahlreiche weitere angeborene und erworbene Stoffwechselstörungen sind denkbar, die zu Mastzellaktivierung führen können. Die Forschung auf diesem Gebiet hat erst gerade begonnen. Ein Beispiel: [Fang et al. 2012].
Nur bei einem sehr kleinen Teil der Systemischen Mastzellaktivierungserkrankungen findet man die oben geschilderten KIT-Mutationen als eine bekannte Ursache und kann sie auf Grund dessen als Mastozytose-Fälle diagnostizieren. Bei den allermeisten Fällen ist es bisher noch nicht gelungen, verantwortliche Genmutationen zu identifizieren. Trotzdem muss irgendeine Störung vorliegen, welche die Mastzellen aktiviert oder leichter aktivierbar macht. Diese Fälle von idiopathischer (=noch unbekannter) Mastzellaktivierung, welche die Diagnosekriterien für eine Mastozytose nicht zu erfüllen vermögen, bezeichnet man als idiopathisches Systemisches Mastzellaktivierungssyndrom (MCAS).
")
Schematische Unterteilung der Systemischen Mastzellaktivierungserkrankungen (MCAD): Bei einer kleinen Zahl von Fällen kann man eine bekannte körperliche Ursache finden, welche zu primärer Mastzellaktivierung führt. Dabei handelt es sich entweder um die seltene Systemische Mastozytose oder um eine äusserst seltene Mastzellleukämie. Die weitaus meisten Fälle bezeichnet man auf Grund der noch unbekannten Ursache als idiopathisches (oder womöglich auch durch ungünstige Umweltfaktoren verursachtes) Mastzellaktivierungssyndrom (MCAS). Man kann bisher nur vermuten, dass es sich möglicherweise ebenfalls um eine primäre Mastzellaktivierung handeln könnte. Nicht zu den MCAD gehören Erkrankungen, die bloss sekundär zu Mastzellaktivierung führen, wie z.B. die Allergien.
Auswirkungen aktivierender Gendefekte:
Verhaltensänderungen aktivierter Mastzellen
Wenn eine Mastzelle aktiviert wird, kann sie folgende Veränderungen erfahren:
- Erleichterte Freisetzung und vermehrte Neusynthese von Mastzellmediatoren
- Aktive Wanderung durch das Gewebe
- Zelldifferenzierung (d.h. Spezialisierung auf bestimmte Funktionen)
- Langlebigkeit durch gestörte Apoptose (=programmierter Zelltod). Dass sie nicht mehr zum geplanten Zeitpunkt eliminiert werden, erhöht deren Anzahl.
- Bei bestimmten Mutationen ist auch eine verstärkte Vermehrung (Proliferation, Zellteilung) der Mastzellen zu beobachten. Dies führt zur Anreicherung krankhaft veränderter Mastzellen in den betroffenen Organen oder Geweben, was zu einer Vergrösserung befallener Organe führen kann, die bei einer Ultraschalluntersuchung sonographisch sichtbar ist.
Je nach körperlicher Ursache und auslösendem Reiz müssen nicht immer alle diese Effekte auftreten. Die Langlebigkeit durch gestörte Apoptose und die beschleunigte Vermehrung führen zu einer langsamen Anreicherung mutierter Mastzellen in den betroffenen Organen bzw. Geweben.
Signalausbreitung, Informationsübertragung auf Zielzellen
Sinn und Zweck der Freisetzung hunderter verschiedener Mastzellmediatoren ist es, mit diesen Botenstoffen biochemische Signale an andere Zellen weiter zu geben. Die umliegenden Gewebezellen, die Zellen des Immunsystems und auch andere Mastzellen sollen informiert werden, was los ist und was in der aktuellen Situation zu tun ist.

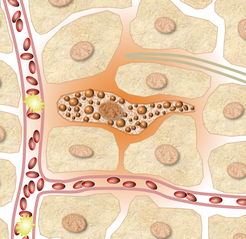
Schematische Zeichnung: Mastzellen im Gewebe, mit Blutkapillaren und Lymphkanal. Die Mastzelle im linken oberen Bereich ist aktiviert und setzt Mediatoren in die Zellzwischenräume frei (orange eingefärbt).
Eine Überaktivierung kann zunächst lokal zu Symptomen führen, indem die umliegenden Zielzellen (normale Körperzellen) durch die ausgeschütteten Mediatoren in einen Ausnahmezustand versetzt werden.
Vor allem werden aber durch die ausgeschütteten Mediatoren auch die noch gesunden Mastzellen in der Umgebung angelockt und ebenfalls aktiviert (sekundäre Aktivierung), wodurch diese ebenfalls beginnen, Mediatoren auszuschütten [Homann et al. 2010, S.545; Homann et al. 2010b, S.194]. Dies aktiviert wiederum noch mehr Mastzellen in noch grösserem Umkreis. Ab einer gewissen Signalstärke können die Mediatoren bis ins Lymphsystem und in die Blutbahn gelangen. Dies mobilisiert und aktiviert wiederum noch mehr Mastzellen in noch grösserer Entfernung. Auf diese Weise kann sich der Krankheitszustand mit der Zeit auf den gesamten Körper ausbreiten. So kann eine lokale Mutation zu einer systemischen (=den gesamten Körper betreffenden) Erkrankung führen. Siehe Abbildungen oben und unten.
Bei sehr ungünstigen Verlaufsformen werden auch Nicht-Mastzell-Immunzellen mit stimuliert. Bei einer solchen immunsystemübergreifenden Erkrankung zeigt eine mastzellspezifische Medikation oft keinen Erfolg mehr.

Diese schematische Zeichnung zeigt eine krankhaft veränderte Mastzelle (links), welche Mediatoren in den Zellzwischenraum (Bildmitte) ausschüttet. Diese Mediatoren wirken nicht nur direkt auf benachbarte Gewebezellen (unten rechts) ein, sondern auch indirekt, indem gesunde Mastzellen (oben rechts) aktiviert werden. Deren freigesetzte Mediatoren docken dann an die Rezeptoren weiterer benachbarter Gewebezellen an.
Bei aktivierten Mastzellen werden aber nicht nur gespeicherte Mediatoren freigesetzt. Auch die Neusynthese von Arachidonsäuremetaboliten (Prostaglandin D2, Leukotriene C4, ...), von Cytokinen und Chemokinen wird angeregt [Akin et al. 2010, S.1100].
Der Verlauf und Schweregrad der Erkrankung wird möglicherweise gar nicht so sehr durch die Art der Mutation bestimmt. Entscheidender sind vermutlich der Reifegrad und Ort der mutierten Zelle und in welche Organe und Gewebe die mutierten Tochterzellen wandern.
Symptome werden ausgelöst durch 1) die freigesetzten Mediatoren und 2) bei den aggressiveren Formen selten auch durch gestörte Organfunktionen bei grossen Mastzellansammlungen in den infiltrierten Geweben, auf Grund von Verdrängung (z.B. Blutarmut (Anämie) bei Knochenmarkinfiltration) [Homann et al. 2010, S.545].
Die Menge ausgeschütteter Mediatoren in einem bestimmten Organ oder Gewebe ist abhängig von:
- Anzahl der (krankhaft veränderten) Mastzellen (Zelldichte). Diese nimmt lokal zu, indem ...
- ... bei aktivierten Mastzellen die Apoptose (programmierter Zelltod) deaktiviert wird. Die Mastzellen werden nicht mehr nach Erreichen der vorgesehenen Lebensdauer eliminiert.
- ... Mastzellen aus anderen Körperteilen durch die Botenstoffe aktivierter Mastzellen chemotaktisch angelockt werden,
- ... pathologisch veränderte Zellen sich unkontrolliert vermehren (was nur bei bestimmten Mutationen der Fall ist).
- Signalverstärkung. Auch die gesunden Mastzellen können sekundär durch die von den mutierten Mastzellen ausgeschütteten Mediatoren alarmiert und aktiviert werden. [Homann et al. 2010, S.545; Homann et al. 2010b, S.194]
- Strukturelle Veränderungen mutierter Mastzellen, die zu verstärkter Freisetzung führen
- Stimulation durch Triggerfaktoren (Aktivierung). Die uns bekannten Auslöser nennen wir auf der Seite Therapie > Auslöser meiden.
Krankheitsverlauf, Prognose
Die Erkrankung kann sowohl plötzlich ausbrechen wie auch ganz allmählich zunehmen oder seit Geburt bestehen.
Das Vorhandensein einer aktivierenden Mutation ist wohl eine notwendige Voraussetzung für das Entstehen der primären MCAD, reicht aber alleine nicht aus. Es gibt Personen, die trotz solcher Mutationen bis an ihr natürliches Lebensende nicht mehr als eine kleine Befindlichkeitsstörung entwickeln. Folglich muss es noch andere Faktoren geben, welche die Aktivierung voran treiben. Das sieht man auch daran, dass es vielen Betroffenen so vorkommt, als bräche die Krankheit von einem Tag auf den anderen plötzlich aus, während man vorher symptomfrei war. Dies ist meist nach besonders belastenden Ereignissen der Fall, z.B. ein schwerer Verkehrsunfall mit Operationen und Spitalaufenthalt. Das kann man vermutlich so erklären, dass die sehr langsame Zunahme mutierter Mastzellen vorher nicht bemerkt wurde, weil sie erst sehr milde und alltägliche Symptome bewirkte. Erst bei einem aussergewöhnlichen Ereignis, bei dem mehrere sehr starke Triggerfaktoren gleichzeitig einwirken (im erwähnten Beispiel: Verletzungen, Desinfektionsmittel, Schmerzmittel, Narkotika, Röntgenkontrastmittel und andere Medikamente, grosser emotionaler Stress), werden die Mastzellen um ein Vielfaches stärker als sonst aktiviert (vielleicht im Zusammenspiel mit dem Immunsystem?) und lösen einen Schub aus, der nicht so schnell wieder abklingt (oder der vielleicht auf noch ungeklärte Weise zu einer dauerhaften Mastzellaktivierung führt?). Auch Infektionen können auslösende Ereignisse sein.
Die Intensität der Symptome ist oft schwankend. Typischerweise treten zu Beginn nur einzelne Phasen mit Symptomen auf (Schübe, Episoden). Mit der Zeit werden dann die Schübe länger, die Pausen dazwischen kürzer, bis man permanent unter chronischen Symptomen leidet. Andere Krankheitsverläufe sind jedoch ebenfalls möglich.
Die Erkrankung ist grundsätzlich nicht ansteckend, könnte aber unter Umständen bei Organspenden auf den Empfänger übertragen werden.
Prognose im Normalfall
Anders als bei bösartigen Krebsformen gerät bei Mastzellaktivierungserkrankungen die Zellteilung nicht ausser Kontrolle. Der Krankheitsverlauf ist daher in den allermeisten Fällen langfristig gesehen konstant oder über die Jahre gesehen ganz langsam ansteigend (wobei unklar ist, ob der Anstieg körperlich bedingt oder auf Umweltfaktoren zurückzuführen ist). Die Sterblichkeit dürfte in etwa gleich gross sein wie bei der Durchschnittsbevölkerung. Die Prognose ist somit "günstig", wie es der Mediziner auszudrücken pflegt. Die Erkrankung ist allerdings nach derzeitigem Forschungsstand grundsätzlich nicht ursächlich heilbar, sondern lediglich medikamentös und mit dem Meiden der Auslöser beherrschbar. Es gibt einzelne Berichte über plötzliche Heilungen (Spontanremissionen), bei denen jedoch wegen der diagnostischen Schwierigkeiten unklar ist, um welche Art von Erkrankung es sich wirklich handelte.
Anaphylaxie, anaphylaktoide Schocks
Nur ein Teil der Betroffenen ist anfällig für anaphylaktoide Reaktionen nach der Exposition mit mastzellaktivierenden Auslösern oder kann sogar ohne einen erkennbaren bekannten Auslöser anaphylaktoide Schocks erleiden (idiopathische Anaphylaxie). Ein derartiger plötzlicher Kreislaufkollaps kann mit rechtzeitig angewendeten Notfallmedikamenten günstig beeinflusst werden. Im schlimmsten Fall kann ein anaphylaktoider oder anaphylaktischer Schock jedoch zum Tode führen. Betroffene mit einer bekannten Neigung zu Anaphylaxie haben daher im Vergleich zu anderen Betroffenen ein erhöhtes Sterblichkeitsrisiko. Dies besonders dann, wenn die Therapiemassnahmen nicht sorgfältig befolgt werden (z.B. Diätfehler, unverträgliche Medikamente).
Aggressivere Verlaufsformen
Nur bei bestimmten aggressiveren Formen von systemischer Mastozytose ist auch eine verstärkte Zellteilung der Mastzellen zu beobachten. Einzelne Organe könnten heftige Beschwerden verursachen, in ihrer Funktion erheblich beeinträchtigt sein oder geschädigt werden.
Nur äusserst selten (bisher weltweit erst wenige Einzelfälle bekannt) kann die Erkrankung in eine Mastzellleukämie über gehen (rasche Vermehrung von Mastzellen in der Blutbahn), mit einer rasend schnellen Verschlechterung des Befindens, was zumeist innert weniger Monate zum Tode führt. Nur sehr schwer behandelbar. Prognose ungünstig.
Empfohlene Lektüre für Betroffene
Die folgenden wissenschaftlichen Publikationen sind deutsch und frei zugänglich:
| Homann et al. 2010b | Homann J, Homann S, Molderings GJ.: "Bemerkungen zur Begutachtung von systemischen Mastzellerkrankungen". Med Sach 106 5/2010. http://www.aerztekammer-bw.de/10aerzte/20fortbildung/20praxis/65medSach/1005.pdf |
| Hartmann et al. 2009 | Karin Hartmann, Tilo Biedermann, Knut Brockow, Jürgen Grabbe, Hans-Peter Horny, Undine Lippert, Marcus Maurer, Martin Raithel, Ernst Rietschel, Franziska Ruëff, Karl Sotlar: "Leitlinie Mastozytose". Allergologie, Jahrgang 32, Nr. 6/2009, S. 199-213. Allergologie, Jahrgang 32, Nr. 6/2009, S. 199-213. Nicht mehr abrufbar: http://www.awmf.org/uploads/tx_szleitlinien/013-058l_S1_Mastozytose.pdf Nicht mehr abrufbar: http://www.derma.de/fileadmin/derma/pdfs/ll_mastozythose.pdf Neuere Version: https://register.awmf.org/de/leitlinien/detail/013-058 (Übersichtsartikel Mastozytose) |
| Molderings et al. 2005 | Molderings, Gerhard J.; Brüss, Michael; Raithel, Martin; Wilken, Verena; Hartmann, Karin; Brockow, Knut; Wardelmann, Eva; Scheurlen, Christian; Homann, Jürgen: "Systemische Mastozytose als Grund für chronische gastrointestinale Beschwerden: Praxisorientierte Hinweise zu Diagnostik und Therapie". Dtsch Arztebl 2005; 102(24): A-1744 / B-1470 / C-1386 https://www.aerzteblatt.de/pdf.asp?id=47323 |
Zusatzinformationen für Fachpersonen
| Molderings et al. 2014 | Molderings GJ, Homann J, Brettner S, Raithel M, Frieling T: "Systemische Mastzellaktivierungserkrankung: Ein praxisorientierter Leitfaden zu Diagnostik und Therapie" [Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options]. Dtsch Med Wochenschr. 2014 Jul;139(30):1523-34; quiz 1535-8. doi: 10.1055/s-0034-1370055. Epub 2014 May 6. http://www.ncbi.nlm.nih.gov/pubmed/24801454 |
| Pardanani 2012 | Pardanani A.: "Systemic mastocytosis in adults: 2012 Update on diagnosis, risk stratification, and management". Am J Hematol. 2012 Apr;87(4):401-11. doi: 10.1002/ajh.23134. http://www.ncbi.nlm.nih.gov/pubmed/22410759 (Guter Überblick über bisher identifizierte mastzellaktivierende Gene, familiäre Mastozytose, Diagnose, Definitionen.) |
| Valent et al. 2012 | Valent P, Akin C, Arock M, Brockow K, Butterfield JH, Carter MC, Castells M, Escribano L, Hartmann K, Lieberman P, Nedoszytko B, Orfao A, Schwartz LB, Sotlar K, Sperr WR, Triggiani M, Valenta R, Horny HP, Metcalfe DD.: "Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal". Int Arch Allergy Immunol. 2012;157(3):215-25. Epub 2011 Oct 27. http://www.ncbi.nlm.nih.gov/pubmed/22041891 (Propose a global unifying classification of all MC disorders and pathologic MC reactions. This classification includes three types of 'MCA syndromes' (MCASs), namely primary MCAS, secondary MCAS and idiopathic MCAS. MCA is now defined by robust and generally applicable criteria, including (1) typical clinical symptoms, (2) a substantial transient increase in serum total tryptase level or an increase in other MC-derived mediators, such as histamine or prostaglandin D(2), or their urinary metabolites, and (3) a response of clinical symptoms to agents that attenuate the production or activities of MC mediators.) |
| Molderings et al. 2011 | Molderings GJ, Brettner S, Homann J, Afrin LB.: "Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options". J Hematol Oncol. 2011 Mar 22;4:10. http://www.ncbi.nlm.nih.gov/pubmed/21418662 |
| Brockow and Ring 2011 | Brockow K, Ring J.: "Update on diagnosis and treatment of mastocytosis". Curr Allergy Asthma Rep. 2011 Aug;11(4):292-9. http://www.ncbi.nlm.nih.gov/pubmed/21523372 |
| Akin et al. 2010 | Akin C, Valent P, Metcalfe DD: "Mast cell activation syndrome: Proposed diagnostic criteria". J Allergy Clin Immunol. 2010 Dec;126(6):1099-104.e4. Epub 2010 Oct 28. http://www.ncbi.nlm.nih.gov/pubmed/21035176 |
| Homann et al. 2010 | Homann J, Kolck UW, Ehnes A, Frieling T, Raithel M, Molderings GJ.: "Die systemische Mastozytose - Standortbestimmung einer internistischen Erkrankung [Systemic mastocytosis - definition of an internal disease]". Med Klin (Munich). 2010 Aug;105(8):544-53. Epub 2010 Sep 8. http://www.ncbi.nlm.nih.gov/pubmed/20824412 |
Geführter Rundgang: Weiter zur Seite
Mastzellerkrankungen > Histaminstoffwechsel
Quellenangaben
Tipp: Der "zurück"-Button Ihres Browsers bringt Sie zur vorherigen Stelle zurück.
| A | Zurück zur vorherigen Stelle |
|---|---|
| Afrin 2014 | Lawrence B. Afrin: "The Bulk of the Iceberg revealed: Mast Cell Activation Syndrome". Gastvortrag vom 6. August 2014 an der University of Cape Town, Südafrika, ca. ab Minute 0:28:00 des Videos. Nicht mehr abrufbar: http://meeting.uct.ac.za/p4j213xndbs/?launcher=false&fcsContent=true&pbMode=normal |
| Akin et al. 2010 | Akin C, Valent P, Metcalfe DD: "Mast cell activation syndrome: Proposed diagnostic criteria". J Allergy Clin Immunol. 2010 Dec;126(6):1099-104.e4. Epub 2010 Oct 28. https://pubmed.ncbi.nlm.nih.gov/21035176 |
| B | Zurück zur vorherigen Stelle |
| Brockow 2013 | Prof. Dr. K. Brockow: "Mastzellaktivierungssyndrome". Der Hautarzt, February 2013, Volume 64, Issue 2, pp 102-106. http://link.springer.com/article/10.1007%2Fs00105-012-2452-6 |
| Brockow and Ring 2011 | Brockow K, Ring J.: "Update on diagnosis and treatment of mastocytosis". Curr Allergy Asthma Rep. 2011 Aug;11(4):292-9. http://www.ncbi.nlm.nih.gov/pubmed/21523372 |
| C | Zurück zur vorherigen Stelle |
| Chaix et al. 2013 | Chaix A, Arcangeli ML, Lopez S, Voisset E, Yang Y, Vita M, Letard S, Audebert S, Finetti P, Birnbaum D, Bertucci F, Aurrand-Lions M, Dubreuil P, De Sepulveda P.: "KIT-D816V oncogenic activity is controlled by the juxtamembrane docking site Y568-Y570". Oncogene. 2013 Feb 18. doi: 10.1038/onc.2013.12. http://www.ncbi.nlm.nih.gov/pubmed/23416972 |
| F | Zurück zur vorherigen Stelle |
| Fang et al. 2012 | Fang X, Lang Y, Wang Y, Mo W, Wei H, Xie J, Yu M.: "Shp2 Activates Fyn and Ras to Regulate RBL-2H3 Mast Cell Activation following FceRI Aggregation". PLoS One. 2012;7(7):e40566. Epub 2012 Jul 10. http://www.ncbi.nlm.nih.gov/pubmed/22802969 |
| H | Zurück zur vorherigen Stelle |
| Hartmann et al. 2009 | Karin Hartmann, Tilo Biedermann, Knut Brockow, Jürgen Grabbe, Hans-Peter Horny, Undine Lippert, Marcus Maurer, Martin Raithel, Ernst Rietschel, Franziska Ruëff, Karl Sotlar: "Leitlinie Mastozytose". Allergologie, Jahrgang 32, Nr. 6/2009, S. 199-213. Allergologie, Jahrgang 32, Nr. 6/2009, S. 199-213. Nicht mehr abrufbar: http://www.awmf.org/uploads/tx_szleitlinien/013-058l_S1_Mastozytose.pdf Nicht mehr abrufbar: http://www.derma.de/fileadmin/derma/pdfs/ll_mastozythose.pdf Neuere Version: https://register.awmf.org/de/leitlinien/detail/013-058 (Übersichtsartikel Mastozytose) |
| Homann et al. 2010b | Homann J, Homann S, Molderings GJ.: "Bemerkungen zur Begutachtung von systemischen Mastzellerkrankungen". Med Sach 106 5/2010. Nicht mehr abrufbar: http://www.aerztekammer-bw.de/10aerzte/20fortbildung/20praxis/65medSach/1005.pdf |
| Homann et al. 2010 | Homann J, Kolck UW, Ehnes A, Frieling T, Raithel M, Molderings GJ.: "Die systemische Mastozytose - Standortbestimmung einer internistischen Erkrankung [Systemic mastocytosis - definition of an internal disease]". Med Klin (Munich). 2010 Aug;105(8):544-53. Epub 2010 Sep 8. http://www.ncbi.nlm.nih.gov/pubmed/20824412 |
| J | Zurück zur vorherigen Stelle |
| Jarisch 2004 | Jarisch, Reinhart: "Histamin-Intoleranz, Histamin-Intoleranz und Seekrankheit", Thieme-Verlag, 2. Auflage, 2004. ISBN 3-13-105382-8 |
| K | Zurück zur vorherigen Stelle |
| Kofler et al. 2009 | H. Kofler, W. Aberer, M. Deibl, Th. Hawranek, G. Klein, N. Reider und N. Fellner: "Diaminoxidase keine diagnostische Hilfe bei Histaminintoleranz", Allergologie, vol. 32, no. 3, pp. 105–109, 2009. https://www.dustri.com/nc/de/deutschsprachige-zeitschriften/mag/allergologie/vol/jahrgang-32-3/issue/maumlrz-1.html (Nur Abstract kostenlos abrufbar) |
| Kofler et al. 2011 | Lukas Kofler, Hanno Ulmer, Heinz Kofler: "Histamine 50-Skin-Prick Test: A Tool to Diagnose Histamine Intolerance", ISRN AllergyVolume 2011 (2011), Article ID 353045, 5 pages. doi:10.5402/2011/353045. https://pubmed.ncbi.nlm.nih.gov/23724226/, abgerufen am 25.11.2011. |
| L | Zurück zur vorherigen Stelle |
| Lillestol et al. 2010 | Lillestøl K1, Helgeland L, Arslan Lied G, Florvaag E, Valeur J, Lind R, Berstad A.: "Indications for atopic bowel in patients with self-reported food hypersensitivity.". Aliment Pharmacol Ther. 2010 May;31(10):1112-1122. doi: 10.1111/j.1365-2036.2010.04261.x. https://pubmed.ncbi.nlm.nih.gov/20163379 Seronegative gastrointestinale Nahrungsmittelallergien. |
| M | Zurück zur vorherigen Stelle |
| Maintz et al. 2006 | Maintz, Laura; Bieber, Thomas; Novak, Natalija: "Die verschiedenen Gesichter der Histaminintoleranz: Konsequenzen für die Praxis (Histamine Intolerance in Clinical Practice)", Deutsches Ärzteblatt 2006; 103(51-52). https://www.aerzteblatt.de/archiv/53958, abgerufen am 25.08.2009. |
| Molderings et al. 2014 | Molderings GJ, Homann J, Brettner S, Raithel M, Frieling T: "Systemische Mastzellaktivierungserkrankung: Ein praxisorientierter Leitfaden zu Diagnostik und Therapie" [Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options]. Dtsch Med Wochenschr. 2014 Jul;139(30):1523-34; quiz 1535-8. doi: 10.1055/s-0034-1370055. Epub 2014 May 6. https://pubmed.ncbi.nlm.nih.gov/24801454 |
| Molderings et al. 2011 | Molderings GJ, Brettner S, Homann J, Afrin LB.: "Mast cell activation disease: a concise practical guide for diagnostic workup and therapeutic options". J Hematol Oncol. 2011 Mar 22;4:10. http://www.ncbi.nlm.nih.gov/pubmed/21418662 Frei zugänglicher Übersichtsartikel |
| Molderings 2010 | Molderings GJ.: "Mast cell function in physiology and pathophysiology." BIOTREND Reviews 2010; 5: 1–9. Nicht mehr abrufbar: https://www.biotrend.com/download/BTReview_Jan2010_Mastcell.pdf Erklärt die Funktion der Mastzellen. Listen mit Rezeptoren, Rezeptorantagonisten und -agonisten, Mastzellmediatoren, Folgeerkrankungen |
| O | Zurück zur vorherigen Stelle |
| Orfanos 1966 | C. Orfanos: "Mastzelle und Mastzelldegranulation". Klinische Wochenschrift, 15. Oktober 1966, Volume 44, Issue 20, pp 1177-1182. https://link.springer.com/article/10.1007/BF01742094 Elektronenmikroskopische Beobachtungen an Gewebe-Mastzellen unterschiedlicher Herkunft und Vorbehandlung |
| P | Zurück zur vorherigen Stelle |
| Pardanani 2012 | Pardanani A.: "Systemic mastocytosis in adults: 2012 Update on diagnosis, risk stratification, and management". Am J Hematol. 2012 Apr;87(4):401-11. doi: 10.1002/ajh.23134. http://www.ncbi.nlm.nih.gov/pubmed/22410759 (Guter Überblick über bisher identifizierte mastzellaktivierende Gene, familiäre Mastozytose, Diagnose, Definitionen.) |
| R | Zurück zur vorherigen Stelle |
| Redaktion | Empfehlung des Redakteurs dieser Website oder des Autors dieser Seite, welche aus den Erfahrungen und Anschauungen von betroffenen Laien hervorgegangen ist und lediglich unseren aktuellen Stand des Unwissens widerspiegelt. |
| Reese et al. 2012 | Imke Reese, Barbara Ballmer-Weber, Kirsten Beyer, Stephan Erdmann, Thomas Fuchs, Jörg Kleinetebbe, Ludger Klimek, Ute Lepp, Margot Henzgen, Bodo Niggemann, Joachim Saloga, Christiane Schäfer, Thomas Werfel, Torsten Zuberbier, Margitta Worm: "Vorgehen bei Verdacht auf Unverträglichkeit gegenüber oral aufgenommenem Histamin. Leitlinie der Deutschen Gesellschaft für Allergologie und klinische Immunologie (DGAKI), der Gesellschaft für Pädiatrische Allergologie und Umweltmedizin (GPA) und des Ärzteverbandes Deutscher Allergologen (ÄDA)". AWMF 2012 Nicht mehr abrufbar: http://www.awmf.org/uploads/tx_szleitlinien/061-030l_S1_Histaminunverträglichkeit_2012.pdf Neuere Version (31.07.2021): https://register.awmf.org/de/leitlinien/detail/061-030 (Leitlinie zur Diagnose des oralen Histaminsyndroms. Konsensusdokument.) |
| T | Zurück zur vorherigen Stelle |
| Traina et al. 2012 | Traina F, Visconte V, Jankowska AM, Makishima H, O'Keefe CL, Elson P, Han Y, Hsieh FH, Sekeres MA, Mali RS, Kalaycio M, Lichtin AE, Advani AS, Duong HK, Copelan E, Kapur R, Olalla Saad ST, Maciejewski JP, Tiu RV: "Single Nucleotide Polymorphism Array Lesions, TET2, DNMT3A, ASXL1 and CBL Mutations Are Present in Systemic Mastocytosis". PLoS One. 2012;7(8):e43090. Epub 2012 Aug 15. http://www.ncbi.nlm.nih.gov/pubmed/22905207 Mutationen in diversen weiteren Genen nebst der KIT (insbesondere TET2, DNMT3A, and ASXL1) sind vermutlich an der Entstehung von Mastozytose beteiligt. Besonders TET2-Mutationen scheinen mit einer verkürzten Lebenserwartung einher zu gehen. |
| V | Zurück zur vorherigen Stelle |
| Valent et al. 2012 | Valent P, Akin C, Arock M, Brockow K, Butterfield JH, Carter MC, Castells M, Escribano L, Hartmann K, Lieberman P, Nedoszytko B, Orfao A, Schwartz LB, Sotlar K, Sperr WR, Triggiani M, Valenta R, Horny HP, Metcalfe DD.: "Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal". Int Arch Allergy Immunol. 2012;157(3):215-25. Epub 2011 Oct 27. http://www.ncbi.nlm.nih.gov/pubmed/22041891 (Propose a global unifying classification of all MC disorders and pathologic MC reactions. This classification includes three types of 'MCA syndromes' (MCASs), namely primary MCAS, secondary MCAS and idiopathic MCAS. MCA is now defined by robust and generally applicable criteria, including (1) typical clinical symptoms, (2) a substantial transient increase in serum total tryptase level or an increase in other MC-derived mediators, such as histamine or prostaglandin D(2), or their urinary metabolites, and (3) a response of clinical symptoms to agents that attenuate the production or activities of MC mediators.) |
| Valent et al. 2007 | Valent P, Akin C, Escribano L, Födinger M, Hartmann K, Brockow K, Castells M, Sperr WR, Kluin-Nelemans HC, Hamdy NA, Lortholary O, Robyn J, van Doormaal J, Sotlar K, Hauswirth AW, Arock M, Hermine O, Hellmann A, Triggiani M, Niedoszytko M, Schwartz LB, Orfao A, Horny HP, Metcalfe DD.: "Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria." Eur J Clin Invest. 2007 Jun;37(6):435-53. https://pubmed.ncbi.nlm.nih.gov/17537151 Frei zugänglicher Übersichtsartikel |
| Vysniauskaite et al. 2015 | Vysniauskaite M, Hertfelder HJ, Oldenburg J, Dreßen P, Brettner S, Homann J, Molderings GJ: "Determination of plasma heparin level improves identification of systemic mast cell activation disease." PLoS One. 2015 Apr 24;10(4):e0124912. doi: 10.1371/journal.pone.0124912. eCollection 2015. https://pubmed.ncbi.nlm.nih.gov/25909362 Frei zugänglicher Artikel (Plasma heparin level appears more sensitive than the other mediators for detecting systemic MC activity in patients with MCAS. The simple, brief venous occlusion test appears to be a useful indicator of the presence of pathologically irritable MCs, at least in the obstructed compartment of the body.) |
| W | Zurück zur vorherigen Stelle |
| Wöhrl et al. 2004 | Wöhrl S, Hemmer W, Focke M, Rappersberger K, Jarisch R.: "Histamine intolerance-like symptoms in healthy volunteers after oral provocation with liquid histamine.". Allergy Asthma Proc. 2004 Sep-Oct;25(5):305-11. Floridsdorf Allergy Center (FAZ), Vienna, Austria. http://www.ncbi.nlm.nih.gov/pubmed/15603203 (50% von zehn gesunden Frauen ohne Anzeichen von Nahrungsmittelunverträglichkeiten in der Vergangenheit reagierten in einer doppelblinden, placebokontrollierten Studie auf die Gabe von 75 mg Histamin in flüssiger Form mit Symptomen, während keine einzige Person auf das Placebo reagierte. Teilweise traten die Reaktionen zeitlich stark verzögert auf.) |
| Y | Zurück zur vorherigen Stelle |
| Yuzawa et al. 2007 | Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J.: "Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor." Cell. 2007 Jul 27;130(2):323-34. http://www.ncbi.nlm.nih.gov/pubmed/17662946 (Struktur des Tyrosinkinase KIT Rezeptors vor und nach Aktivierung durch den Stammzellfaktor sowie onkogene Mutationen.) |
| Z | Zurück zur vorherigen Stelle |
| Zopf et al. 2009 | Zopf, Yurdagül; Baenkler, Hanns-Wolf; Silbermann, Andrea; Hahn, Eckhart G.;Raithel, Martin: "Differenzialdiagnose von Nahrungsmittelunverträglichkeiten / The Differential Diagnosis of Food Intolerance". Dtsch Arztebl Int 2009; 106(21): 359-69 |